Search
Outsourcing in the Pharmaceutical and Life Sciences Sectors – a UK perspective
Posted
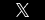


Pharmaceutical and Life Sciences companies operate in a demanding environment and face diverse challenges such as pricing pressure, increased regulatory requirements and mounting costs. With this backdrop, they have, starting with non-core functions, such as IT, facilities management, finance and human resources, before moving to secondary core functions, such as research and development, manufacturing, logistics, warehousing and brokerage, increasingly looked to outsourcing so as to better focus on core competencies, access specialized expertise and achieve cost-saving benefits.
In this blog post, a closer look at some of the key challenges faced by those operating in the Pharmaceutical and Life Sciences sector is taken from an outsourcing perspective.
Regulatory Environment
As might be expected, the Pharmaceutical and Life Sciences sector is subject to an extensive network of rules and regulations. At EU-level, there are a number of European Directives such as Directive 2001/83/EC relating to medicinal products for human use, Directive 2001/20/EC relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use, and Commission Directive 2003/94/EC laying down the principles and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use.
In the UK, the Medicines & Healthcare products Regulatory Agency (MHRA) regulates medicines, medical devices and blood components for transfusion. Its responsibilities include ensuring that medicines etc. meet applicable standards of safety, quality and efficacy and that the supply chain for medicines, medical devices and blood components is safe and secure.
The EU operates a mutual recognition system intended to allow products to move unhindered between national markets—each other member state has an equivalent national competent authority to the MHRA, such as France’s National Agency for the Safety of Medicine and Health Products and Germany’s Federal Institute for Drugs and Medical Devices. The national competent authorities work closely with the European Medicines Agency (EMA) and the European Commission—the Commission’s principal role in the European medicines regulatory system is to make binding decisions based on the scientific recommendations delivered by the EMA and publish guidance defining required good practices.
Consequently, outsourcing and other commercial agreements made by Pharmaceutical and Life Sciences companies must reflect the heavy regulatory burden to which they are subject and will include provisions dealing with topics such as audits and inspections, retention of documents, protection of sensitive and other confidential information and data, adherence to company policies, and compliance with laws and regulations, in addition to schedules which detail the scope of service, the system of performance management (i.e., service levels and service credits) and the applicable commercial model and charging structures. The third party provider’s adherence to and compliance with GxPs (see below) is another key area.
Good X Practice (GxP)
GxP is a general term for good (anything…) practice and refers to applicable quality guidelines and regulations. These guidelines are used in many sectors including pharmaceutical, medical devices/software and food industries—their overall intent is to ensure that products are safe for consumers and to ensure the integrity of data used to make product-related safety decisions. In this context “X” can mean Manufacturing (GMP), Clinical (GCP), Laboratory (GLP), Storage (GSP), Distribution (GDP), Pharmacovigilence Practice (GVP) etc.
Organisations needing to comply with GMP and/or GDP include those holding a manufacturer’s licence, a wholesale dealer licence or a blood establishment authorisation, as well as non-UK sites employed by UK marketing authorisation (MA) holders.
In the context of Pharmaceutical and Life Sciences outsourcing generally, two of the most common good practices are GMP and GDP, as they can apply across a range of outsourced activities and functions such as contract manufacturing, integrated facilities management, logistics, brokerage and warehousing:
- GMP is the minimum standard that a medicines manufacturer’s production processes must meet. Products must (a) be of consistent high quality, (b) be appropriate to their intended use, and (c) meet the requirements of the MA or product specification.
- GDP requires that medicines are obtained from the licensed supply chain and are consistently stored, transported and handled under suitable conditions, as required by the MA or product specification.
In addition to the good practice guides published by the European Commission (see footnotes 1 and 2), the MHRA—as the UK’s national competent authority—publishes its own guidance. As with most regulators, the MHRA updates its guidance from time to time—most recently on 23 May 2017 (an update to the GDP compliance report form).
Inspection and Audit
The MHRA inspects manufacturing and distribution sites for GxP compliance as part of the initial licensing/authorisation process and then periodically. Each manufacturer and wholesaler is given a risk rating or score by the MHRA based on the organisation’s compliance report, previous inspection history and organisational changes. No appeal is permitted, although reasons for the risk rating/score are provided once the inspection has taken place. Inspections of organisations with the highest rating or score are prioritised. The MHRA usually gives prior notice although the short-notice inspection programme means that little or no notification can be given, especially in cases of possible breach (e.g., where a report is received from a whistleblower or another MHRA department or regulator). Usually, however, the likely date of the next inspection is known as the MHRA includes this in its inspection reports.
At the inspection, the inspectors examine the systems used to manufacture and/or distribute medicines. Unless it is a short-notice inspection, the organisation will have completed and submitted to the MHRA a compliance report beforehand. The inspection team will interview relevant personnel, review documents and conduct site visits. Site visits may cover any facility or process involved in the production, purchase and distribution of medicines. Key areas likely to be inspected include:
- manufacturing areas;
- quality control (QC) laboratories;
- stock and stock management;
- storage areas;
- temperature monitoring;
- returns areas;
- purchasing and sales functions; and
- transportation arrangements.
Inspections can sometimes be carried out with other MHRA inspections such as good clinical practice or good pharmacovigilance practice. Product-related inspections can also be requested by the EMA. Where any function covered by the above scope has been outsourced to a third party provider, it is vitally important that the MHRA has the exact same access to the provider and its facilities and personnel.
Types of deficiencies
Deficiencies found during inspections are graded at 3 levels—critical, major and other. These are defined in the “Compilation of Community Procedures on Inspections and Exchange of Information” published by the EMA. (See page 47.)
| Type of Deficiency | Definition | Example |
| Critical Deficiency | Any departure from Guidelines on Good Distribution Practice resulting in a medicinal product causing a significant risk to the patient and public health. This includes an activity increasing the risk of falsified medicines reaching the patients.
A combination of a number of major deficiencies that indicates a serious systems failure. |
Examples given by the EMA:
Purchase from or supply of medicinal products to a non-authorised person. Storage of products requiring refrigeration at ambient temperatures. Rejected or recalled products found in sellable stock. |
| Major Deficiency | A non-critical deficiency which:
– a major deviation from Good Distribution Practice; – has caused or may cause a medicinal product not to comply with its marketing authorisation in particular its storage and transport conditions; or – indicates a major deviation from the terms and provisions of the wholesale distribution authorisation. A combination of several other deficiencies, none of which on their own may be major, but which may together represent a major deficiency. |
No examples of major deficiencies are given by the EMA. However, the MHRA report on 2016 GMP inspections cited 449 major deficiencies in quality systems (in this category there were 38 critical and 772 other deficiencies). The next highest number of major deficiencies were in the categories of sterility assurance and production (also the second and third highest categories for critical deficiencies).
|
| Other Deficiency | A deficiency which cannot be classified as either critical or major, but which indicates a departure from Guidelines on Good Distribution Practice. | No examples of other deficiencies are given by the EMA. However, a deficiency may be classified as “other” because it is judged as minor or because there is insufficient information to classify it as major or critical. |
Pharmaceutical and Life Sciences companies contemplating outsourcing should design their performance management systems in the light of the above, with robust processes and remedies particularly in the event of any Critical or Major Deficiency attributable to the third party provider. Remedies may include service credits, corrective action and other remediation, and ultimately termination.
Technical/Quality Agreements
Technical Agreements—also known as Quality Agreements—are required wherever an outsourced activity is covered by applicable good practice Guides (e.g., GMP or GDP). In the case of GMP, the applicable EU rules relating to outsourcing are found in Chapter 7 of the EU GMP Guide which provides:
“Outsourced activities must be correctly defined, agreed and controlled in order to avoid misunderstandings which could result in a product or operation of unsatisfactory quality. There must be a written contract between the Contract Giver and the Contract Acceptor which clearly establishes the duties of each party…”
“Technical aspects of the contract should be drawn up by competent persons suitably knowledgeable in related outsourced activities and Good Manufacturing Practice.”
Other requirements include:
- a contract which covers all technical and other arrangements for the outsourced activities (and the related products or operations);
- adherence to applicable regulations and the Marketing Authorisation for the in-scope product(s);
- ultimate responsibility of the Contract Giver (i.e., the customer) for ensuring that its pharmaceutical quality system covers control and review of the outsourced activities and that adequate processes are in force;
- clear definition of the responsibilities of both parties (i.e., the Contract Giver and the Contract Acceptor (i.e., the third party provider)), clearly stating who undertakes each step of the outsourced activity:
- knowledge management;
- technology transfer;
- supply chain and subcontracting;
- quality and purchasing of materials;
- testing and releasing materials; and
- undertaking production and quality controls (including in-process controls, sampling and analysis);
- documented communication processes between the parties relating to the outsourced activities;
- access to records (including in case of invocation of the documented defect procedures) and applicable document retention requirements; and
- rights to audit the Contract Acceptor and any approved subcontractors.
Getting the Technical Agreement right
This is important. The Technical Agreement spells out the GxP responsibilities of each of the parties, their communication and assurance processes and will nearly always be reviewed by the MHRA (or indeed any other applicable regulator such as the U.S. Food and Drug Administration). The MHRA’s 2016 deficiency report gives the following example of deficiencies related to Technical Agreements sampled by them in the period.
Similar rules are set out in Chapter 7 of the GDP Guide. The ICH Good Manufacturing Practice Guide also requires a Technical Agreement in the context of the contract manufacture of APIs (active pharmaceutical ingredients).
| Deficiency | MHRA Example |
| Insufficiently detailed | The Technical Agreement between Company A and Company B was insufficiently detailed. It only contained a series of bullet points covering Company B’s activities, and did not describe the responsibilities of Company A. |
| Unclear lines of responsibility | The Technical Agreement between Company A and Company C contained conflicting statements regarding the responsibility for customer verification. |
| Scope not described | The Technical Agreement with Company D did not identify the products that were to be within the scope of the agreement. |
| Status of parties unclear | The Technical Agreement with Company E did not identify which party was the Contract Acceptor and which was the Contract Giver. |
| No express requirements | There was no explicit requirement in the Technical Agreement for temperature monitoring devices to be used for shipment of goods to Company F. |
Relationship with outsourcing and other commercial agreements
GxP compliance requires clear, accurate and detailed Technical Agreements to ensure that the Contract Acceptor complies with applicable standards and technical requirements such as storage conditions, stock control and temperature monitoring. In the context of an outsourcing transaction or other commercial arrangement (such as a long term supply agreement), the Technical Agreement will sit alongside the outsourcing/commercial agreement. They are not standalone documents—each should reference the other since they relate to the same set of activities but address different aspects of the relationship between the Contract Giver and the Contract Acceptor. It is important to ensure that the two documents work in concert with and are consistent with each other, and the relationship between the two agreements is clear (i.e., what happens if there is a contract breach and how are any limits on liability determined). Since template Technical Agreements often contain provisions which would typically be contained in the outsourcing / commercial agreement such as dispute resolution, change control and audit/inspection, care needs to be taken so that there is no overlap or conflict between them, ideally by removing any duplication or overlap.
Other points to watch include ensuring that the parties to the outsourcing/commercial agreement are the same as those to the Technical Agreement—if they are not (i.e., the third party provider’s function undertaking the quality-related aspects of an outsourced service resides in a different group entity to the primary provider, then address this through appropriate subcontracting provisions in the outsourcing/commercial agreement. The two agreements should also be co-terminous—the Technical Agreement doesn’t need to contain termination provisions, but should simply come to an end at the same time as the outsourcing/commercial agreement. Finally, the Technical Agreement should not contain any of the commercial terms (service levels, pricing, etc.) nor should it deal with legal terms such as confidentiality, warranty, indemnity and liability—all of which should be handled in the outsourcing/commercial agreement and its schedules.
Brexit
It seems unlikely Brexit will have significant impact on outsourcing of GxP activities by UK-headquartered Pharmaceutical and Life Sciences companies from a GxP compliance perspective—in other words the need to comply will continue, albeit additional requirements will entail since technical speaking, from an EU viewpoint, the UK will become a third country from the stroke of midnight on 30 March 2019 (unless an extension is agreed by the UK and the EU27 in the forthcoming negotiations).
In a recently published Q&A, the European Commission made clear the UK-based manufacturers of APIs will be treated just the same as Chinese, Indian and other third country based manufacturers. For example, the export of APIs from the UK to the EU will require written confirmation from the “competent authority of the exporting third country” in order to verify a plant has been inspected and that its processes are up to the EMA standards. Alternatively, the UK may be able to negotiate an exception (Switzerland has had one since 2012) based on an equivalency finding by the European Commission.